

收稿日期:2023-08-24
DOI:10.19850/j.cnki.2096-4706.2024.05.035
摘" 要:预测药物与其靶向蛋白的结合亲和力是研发新药的关键步骤。传统的湿实验耗时长,成本高。随着人工智能技术的快速发展,在药物筛选阶段应用深度学习的技术可以大幅度提升研发效率。针对上述问题,提出一种基于卷积神经网络预测药物靶点亲和力的方法。将蛋白质和小分子的结构特征分别转换成对应的三维矩阵,送入对应的三维卷积神经网络中进行训练,然后再通过若干层全连接神经网络提取特征值,得到最终的亲和力值。实验结果表明,该模型可有效地预测药物靶点亲和力,具有良好的应用前景。
关键词:人工智能;深度学习;卷积神经网络;蛋白质结构;药物靶点亲和力预测
中图分类号:TP39;T18" " 文献标识码:A 文章编号:2096-4706(2024)05-0162-05
Prediction of Drug Target Binding Affinity Based on Structural Features
SHAO Yunchang, ZHANG Yuanyuan, JIANG Mingjian
(Qingdao University of Technology, Qingdao" 266520, China)
Abstract: Predicting the binding affinity between drugs and their target proteins is a key steps in developing new drugs. Traditional wet experiments are time-consuming and expensive. With the rapid development of artificial intelligence technology, the application of Deep Learning technology in the drug screening phase has the potential to significantly enhance research and development efficiency. A method for predicting drug target binding affinity based on Convolutional Neural Networks is proposed to address the above issues. The structural features of proteins and small molecules are transformed into corresponding three-dimensional matrices, these matrices are fed into respective three-dimensional Convolutional Neural Networks for training. Then, feature values are extracted through several layers of fully connected neural networks to obtain the final binding affinity value. The experimental results indicate that the model can effectively predict the binding affinity of drug targets and has good application prospects.
Keywords: Artificial Intelligence; Deep Learning; Convolutional Neural Networks; protein structure; prediction of drug target binding affinity
0" 引" 言
一款新药物的研发需要投入大量的时间、昂贵的成本,且成功概率低。如在美国,新药的研发大概需要投入26亿美元[1],并且需要17年的时间才能获得美国食品及药物管理局(FDA)的批准[2,3],因此找到一种新的方式来提升药物研发的效率成为当前的迫切需求。
药物靶点的相互作用是一种二元分类的问题,我们认为药物与靶点连续的亲和力的值能够更加直观、准确地反映出二者的结合程度,因此预测药物与靶点的结合亲和力是研发新药的关键步骤,将结合亲和力强的药物靶点筛选出来进行湿实验,可大幅度提升研发效率。借助计算机来预测结合亲和力成为当前比较重要的一种方式。传统的机器学习有RF-Score [4],其构建完全依赖于数据,借助非参数机器学习方法巧妙地规避了对存在问题的建模假设的需求。
随着进入大数据时代以及计算机算力的高速发展,深度学习在图像识别、自然语言处理等应用中获取了巨大成就,越来越多的方法也将药物靶点亲和力预测的问题用深度学习来处理。药物小分子和蛋白质分别都有两种特征表达方式,一种是基于序列的特征,一种是基于结构的特征。基于序列特征处理的深度学习方法比较有代表性的是DeepDTA [5]、GraphDTA [6]。DeepDTA将蛋白质序列和小分子的SMILES序列的字符分别用不同的数字表示,经过嵌入层处理后,将小分子、蛋白质分别送入相应的一维卷积神经网络中训练,预测亲和力值。GraphDTA根据小分子的特性首次将SMILES序列处理成图的形式,并将小分子图送入到图神经网络中训练。比起基于序列特性的方法,基于结构特性的方法在预测上往往更加准确。如DeepSite [7]基于蛋白质结构的信息根据距离和体积重叠的方法进行了蛋白质配体结合位点的预测。KDEEP [8]使用DeepSite的思想并结合卷积神经网络来预测药物靶点结合亲和力。
在本文中,我们提出了一种新颖的基于蛋白质和小分子结构的方法来预测药物靶点结合亲和力。根据蛋白质和小分子的结构文件,分别将其网格化为两个三维特征矩阵,再分别用两个三维卷积神经网络训练两个三维特征矩阵,最终得到结合亲和力的值。实验结果表明我们提出的方法是可拓展、可优化的,而且适用于任何已知三维结构的蛋白质和小分子。
1" 处理方法
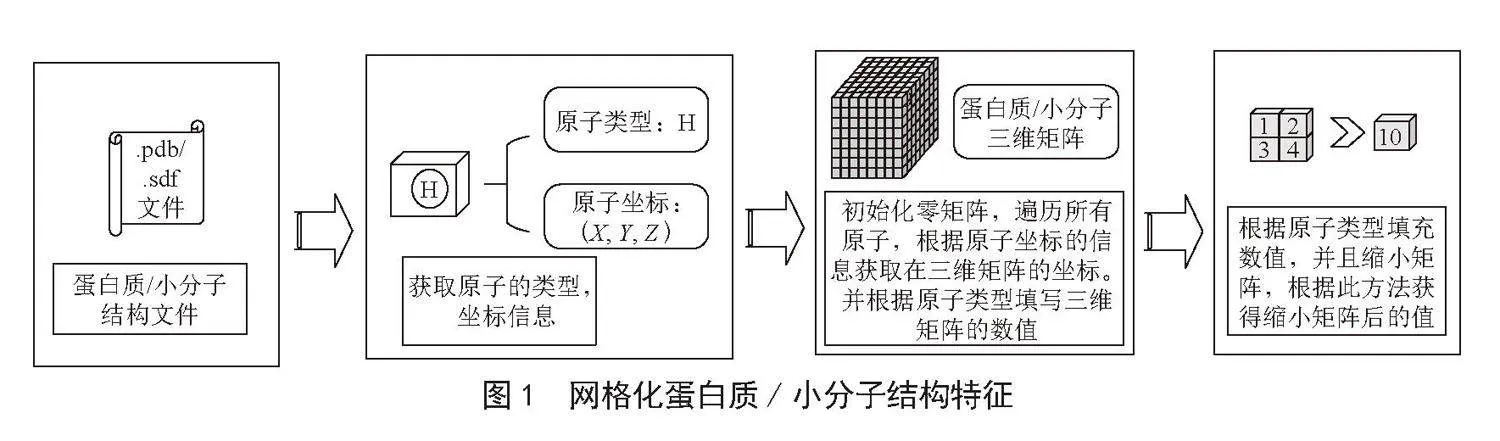
本节介绍关于蛋白质小分子特征处理方式。根据蛋白质的.pdb文件和小分子的.sdf文件,我们可以获取到蛋白质和小分子的结构特征信息,初始化分别代表蛋白质和小分子的零矩阵,为了防止坐标越界,蛋白质或小分子零矩阵的大小根据数据集中蛋白质或小分子坐标的最大X轴值、Y轴值、Z轴值和最小X轴值、Y轴值、Z轴值的差值来定,具体内容见小节4.2。通过小节1.1和小节1.2的方法分别将蛋白质和小分子网格化成两个三维矩阵,具体操作见图1所示。
1.1" 蛋白质的表征方式
根据蛋白质的.pdb文件,我们使用Biopython获取蛋白质结构信息。通过Biopython中提供的方法,遍历蛋白质的所有原子(atom),获取到每个原子的三维坐标信息、原子类型信息。根据原子的坐标信息,我们获取到X轴、Y轴以及Z轴的最小值(X,Y,Z),并以此为原点得到蛋白质三维矩阵的坐标。如原子用Biopython中atom.get_coord()方法获取的坐标为(X1,Y1,Z1),则在我们构建的三维矩阵的坐标为(X1-X,Y1-Y,Z1-Z),遍历所有的蛋白质原子,得到其在蛋白质三维矩阵中的坐标信息。三维矩阵的值是使用Biopython中的atom.element获取原子类型,根据原子类型的不同,从1开始赋予不同的值,如{FE:1,NI:2,H:3},以此类推填写初始化零矩阵的值。我们将得到的三维矩阵的尺寸缩小1 000倍,如若矩阵多个点合成一个,那么矩阵中的数值在缩小的过程中合并相加,得到最终的蛋白质三维矩阵。
1.2" 小分子的表征方式
小分子的处理方式和1.1小节中蛋白质处理的方式大致相同。根据小分子的.sdf文件来获取小分子结构的坐标信息和类型信息。根据小分子的坐标信息,我们获取到X轴,Y轴以及Z轴的最小值(X,Y,Z),并以此为原点得到小分子三维矩阵的坐标。如我们获取的坐标是(X2,Y2,Z2),则小分子的三维矩阵中的坐标是(X2-X,Y2-Y,Z2-Z)。遍历所有的小分子原子,得到其在小分子三维矩阵中的坐标信息。三维矩阵的值根据原子类型的不同,从1开始赋予不同的值,如{Br:1,F:2,C:3},以此类推填写初始化零矩阵的值。我们将得到的三维矩阵的尺寸缩小8倍,矩阵中的数值与小节1.1中蛋白质矩阵同样的处理方式,得到最终的小分子三维矩阵。
2" 神经网络
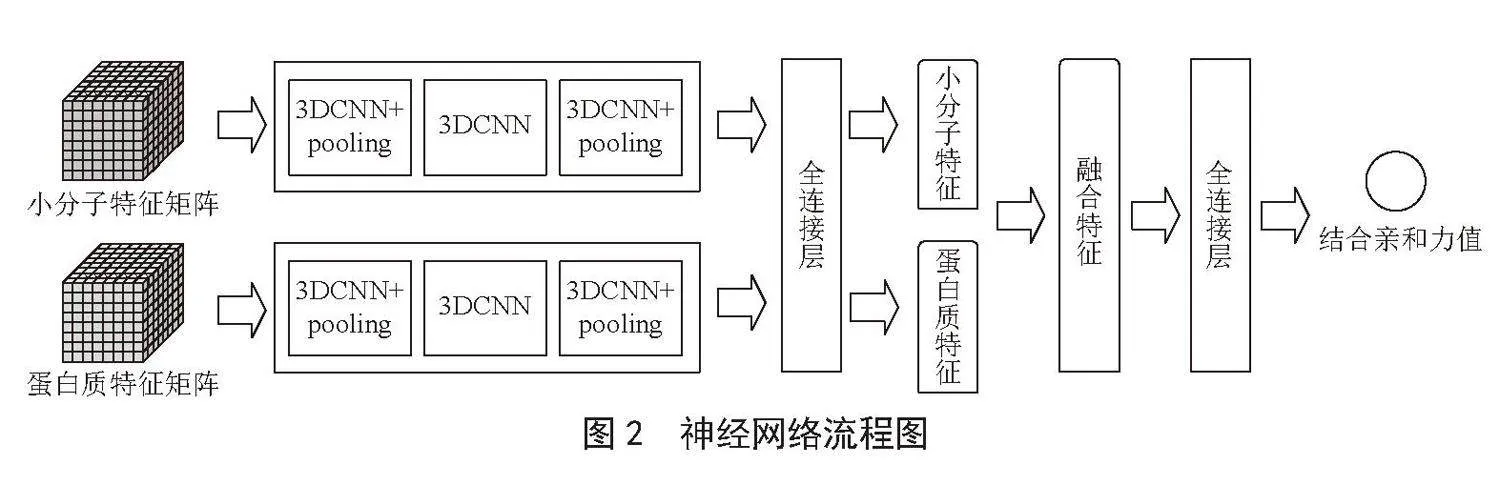
小节1中我们介绍了关于结构特征的处理方式,将蛋白质和小分子结构网格化,形成了两个三维特征矩阵。这一节我们介绍处理两个三维特征矩阵的神经网络的结构,以及提取特征的步骤。我们将两个三维特征矩阵分别送入对应的三维卷积神经网络中提取特征,将获得的特征平铺后分别送入两个全连接层进一步提取特征,得到两个分别代表蛋白质和小分子的128维向量,将两种向量进一步融合为256维的向量,最后经过全连接层得到一维的向量输出值。2.1和2.2小节详细介绍三维卷积神经网络和全连接神经网络的结构。具体的神经网络流程如图2所示。
2.1" 三维卷积神经网络
首先使用三层三维卷积神经网络分别卷积蛋白质的三维矩阵和小分子的三维矩阵。每层卷积神经网络的卷积核大小都是(3,3,3),为了防止矩阵边缘信息在卷积过程中缺失,每层卷积神经网络我们都加大小为(2,2,2)的填充(padding),由于神经网络的输入是一个蛋白质矩阵或者小分子矩阵,所以第一层的三维卷积神经网络的输入通道是1,输出通道是16;第二层的三维卷积神经网络的输入通道是16,输出通道是32;第三层的三维卷积神经网络的输入通道是32,输出通道是1。为了特征提取更加精准,我们还引入了最大池化层(Maxpooling),池化层的卷积核为(2,2,2)。每层卷积后使用的激活函数是ReLU [9]。
2.2" 全连接层
经过上述的三维卷积层后,我们得到了分别代表蛋白质和小分子特征值,将特征值平铺后我们将其分别送入两个三层的全连接层。第一层将输入的向量变为2 048维度,第二层由2 048维度变为1 024维度,第三层由1 024维度变为128维度。激活函数使用的是ReLU,同时为了防止过拟合,我们还引入了Dropout层。至此我们获得了分别代表蛋白质和小分子的128维向量。将两个128维向量融合成一个256维的特征向量送入下一个三层全连接层进行训练,第一层特征向量由256维变成1 024维度,第二层由1 024维度变成512维度,最后一层得到的输出是一个一维值,也就是我们所需要的亲和力的特征表示。
3" 数据集及衡量标准
这一章节我们介绍实验所用到的数据集和衡量实验性能的公式。3.1小节介绍PDBbind数据集[10],3.2小节介绍了本文实验所使用的数据集划分,3.3小节介绍我们对于实验所使用的衡量标准。
3.1" PDBbind数据集的介绍
PDBbind作为一个与蛋白质-配体相互作用相关的数据库,在测量解离常数(Kd)、抑制常数(Ki)或半浓度(IC50)等物理量以衡量相互作用强度的基础上,为药物设计、分子对接、虚拟筛选等计算生物学研究提供了必要的支持。在当前研究中,PDBbind展现出其作为实验方法测试平台的卓越选择。PDBbind数据库包含三个主要子集:通用集、精炼集和核心集。通用集包含大量蛋白质-配体复合物数据,囊括了多样性的结构与相互作用模式。该子集广泛涵盖多种复合物类型,为研究者提供多样性数据样本。精炼集是通用集的子集,经过严格筛选以保留高质量数据,其所蕴含的结构与相互作用信息更为可靠。而核心集则是精炼集的更为精选子集,汇聚了代表性的蛋白质-配体复合物数据。这些复合物在结构生物学领域具备显著的意义,能够作为研究特定领域问题的基准数据。
3.2" 数据集的划分
在本文中我们进行的实验使用到PDBbind2016。在PDBbind2016中精炼集包含4 057组数据,核心集包含290组数据,对于实验标签Kd、Ki、IC50,我们都根据Kd的处理方式,如式(1)所示。为了防止过拟合现象,我们要确保在训练集和测试集中不能出现同一组数据,所以我们将在精炼集出现的核心集的数据全部删除,得到3 767组数据,将这3 767组数据作为实验的训练集,290组数据作为实验的测试集。
(1)
3.3" 衡量标准
在本文中,我们使用了均方根误差(RMSE),皮尔森相关系数(R),斯皮尔曼等级相关系数(Rs)来衡量我们的实验性能。以下是关于各个公式的详细介绍。
3.3.1" 均方根误差RMSE
RMSE用于衡量预测模型的性能,因此它的计算涉及预测值与实际观测值之间的差异,RMSE的数值越小,说明模型的准确率越高。RMSE的计算式如下:
(2)
式中n表示数据点的总数;yi表示实际观测值; 表示对应的预测值。
3.3.2" 皮尔森相关系数R
皮尔森相关系数R被广泛应用于衡量两个连续变量之间线性关系的强度和方向,其取值范围被限制在-1到1之间。具体而言,当R趋近于1时,暗示着两变量之间存在着完全正向线性关系,即其中一个变量的增加伴随着另一个变量的严格增加。相反地,当R趋近于-1时,意味着两个变量之间呈现明显的负向线性关系,即一个变量的增加伴随着另一个变量的严格减少。然而,当R接近于0时,它表明两变量之间的线性关系较为弱化,或者还存在其他可能的非线性关系。皮尔森相关系数在分析变量关联性方面具有突出的价值,帮助研究者洞察变量之间的态势与相互联系,从而为更深入的分析提供了基础。R的计算式如下:
(3)
式中xi和yi分别表示样板第i个观测值, 和" 分别表示x和y的均值。
3.3.3" 斯皮尔曼等级相关系数Rs
斯皮尔曼等级相关系数(Rs)是一项重要的统计工具,其用途在于测量两个变量之间的关联性。与先前提及的皮尔逊相关系数相异,Rs并不对变量间呈线性关系提出要求,而是以变量的等级或顺序数据为基础进行分析。Rs的取值范围界定于区间[-1,1]之内。当Rs等于1时,意味着存在着完全正相关关系,即两变量的秩次排序完全一致;当Rs趋近于0,则意味着变量之间缺乏显著的秩次关联性;而当Rs为-1时,则指示出完全负相关,揭示了两变量秩次排序的完全逆序关系。不容忽视的是,斯皮尔曼等级相关系数不仅适用于分析非线性关系,还对数据集中存在离群值的情形表现出鲁棒性。其所具有的尺度不变性使其能够有效地克服数据尺度变换所引发的问题。如此特性使得Rs在解决无法满足线性关系假设的问题时效果显著。综上所述,斯皮尔曼等级相关系数作为一种统计工具,应用广泛,主要用于测量变量之间的秩次关联性。特别是在处理无法满足线性关系假设的场景下,其优越性愈加显著。Rs的表示计算公式如下:
(4)
式中di表示每对数据点在两个变量中的秩次差,n表示样本数量。
4" 实验及分析
在小节3中我们介绍了关于本文的实验用到的数据集和衡量标准,这一节我们运用小节3的内容进行实验并且详细介绍实验的操作步骤。
4.1" 实验配置及其过程
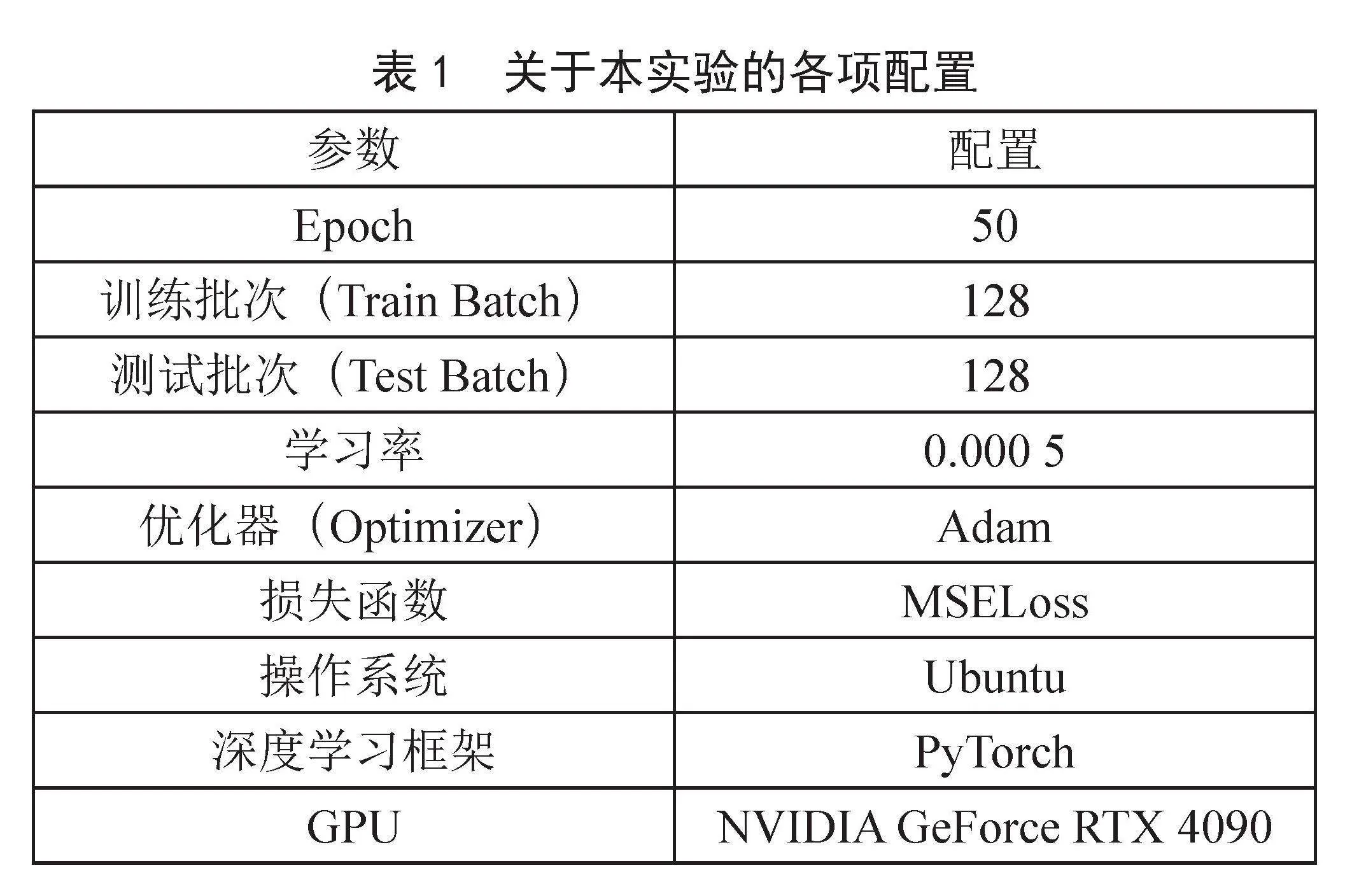
本实验基于Ubuntu操作系统,数据集使用PDBbind2016。为了加快训练速度,我们采用显卡(GPU)进行训练。具体的配置如表1所示。
表1" 关于本实验的各项配置
参数 配置
Epoch 50
训练批次(Train Batch) 128
测试批次(Test Batch) 128
学习率 0.000 5
优化器(Optimizer) Adam
损失函数 MSELoss
操作系统 Ubuntu
深度学习框架 PyTorch
GPU NVIDIA GeForce RTX 4090
首先我们需要将PDBbind数据集转换成PyTorch格式。将.pdb文件和.sdf文件所提供的信息创建成分别代表蛋白质和小分子的三维矩阵,并且将蛋白质、小分子矩阵以及所对应的亲和力的值以三个一组的形式存储起来。原始数据集形成PyTorch格式处理过的训练集和测试集,并保存成两种文件,将训练集文件送到神经网络中训练50轮,并将得到的模型参数用于测试集文件测试。
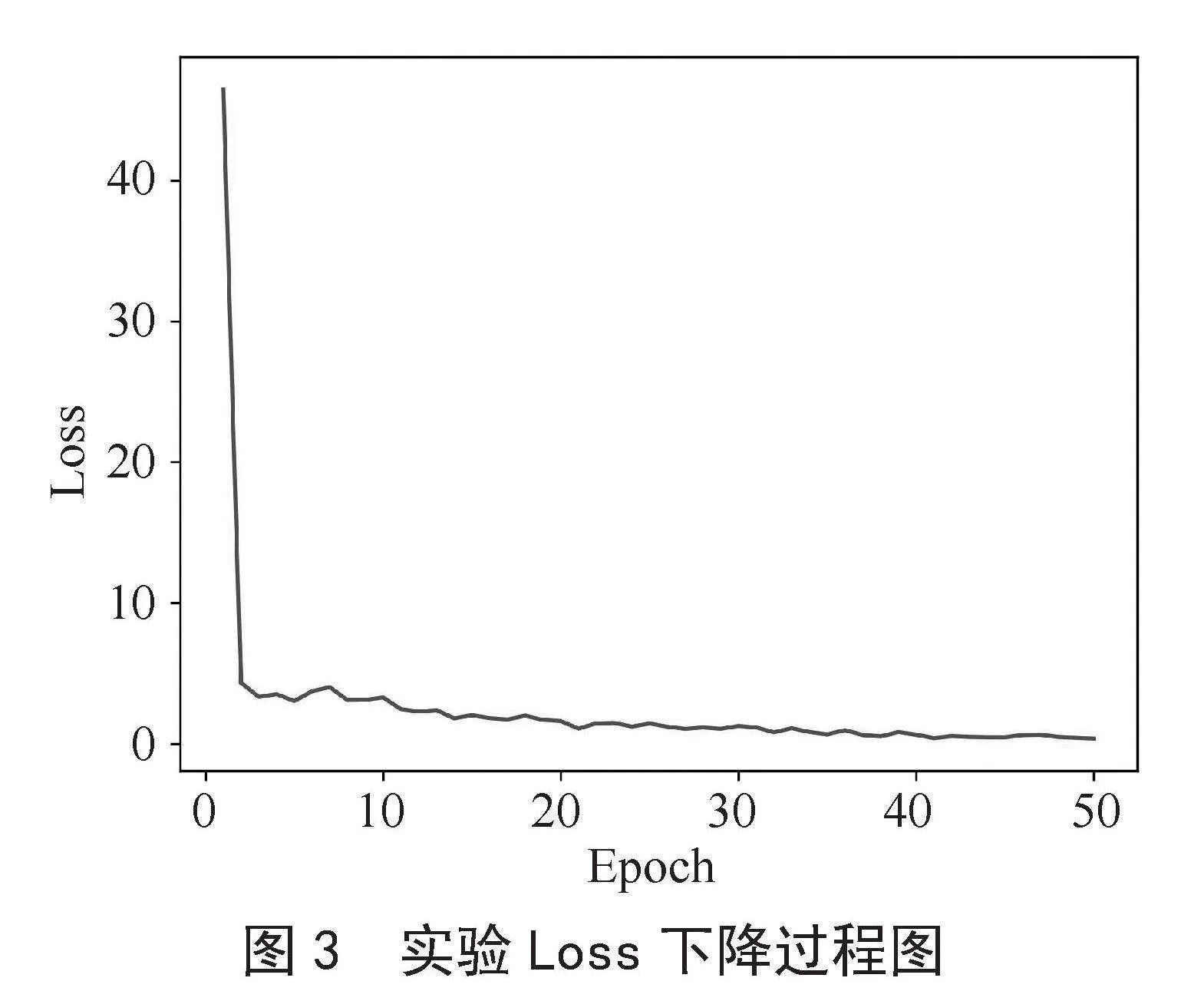
按照上述的实验流程,我们进行一个完整的实验,实验中Loss下降的过程如图3所示。从图中我们可以看出,横坐标训练轮次(Epoch)为50轮,纵坐标Loss的数值随着Epoch的增加而逐步减小。这表明我们所提出的模型,随着训练次数的增加,预测结果与实际结果逐渐接近,可以有效地提升预测的准确率。
图3" 实验Loss下降过程图
4.2" 基于PDBbind2016数据集的实验
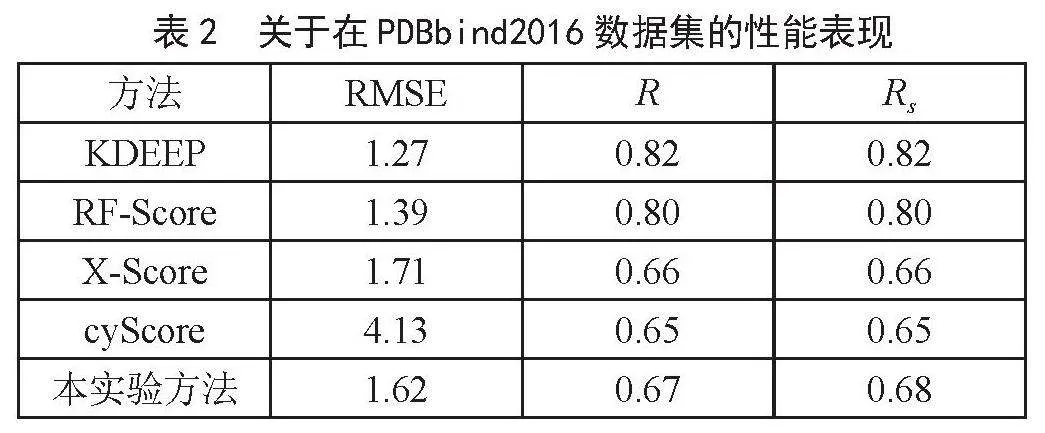
根据PDBbind2016数据集计算蛋白质和小分子的原子分别在X、Y、Z轴上的最大坐标值与最小坐标值的差值,选出数据集中每个维度最大的差值作为初始化零矩阵的对应维度的边长,并且在最外层加以零填充,我们将蛋白质零矩阵的大小定为(170,170,190),小分子零矩阵的大小定为(30,30,30),缩小后蛋白质和小分子矩阵大小分别为(17,17,19),(15,15,15)。我们根据KDEEP的实验操作划分了数据集,并使用KDEEP提供的实验数据,有4种方法与我们的实验方法在RMSE、R、Rs的衡量标准上进行对比,分别是KDEEP [8]、RF-Score [4]、X-Score [11]、cyScore [12],对比的详细结果如表2所示。
表2" 关于在PDBbind2016数据集的性能表现
方法 RMSE R Rs
KDEEP 1.27 0.82 0.82
RF-Score 1.39 0.80 0.80
X-Score 1.71 0.66 0.66
cyScore 4.13 0.65 0.65
本实验方法 1.62 0.67 0.68
从表2中可以看出我们提出的方法RMSE是1.62,R是0.67,Rs是0.68。在RMSE、R、Rs上的表现都优于传统的评分方法X-Score,cyScore,这得益于我们的方法通过网格化结构信息,用数字来代替原子类型,能够更好地表达蛋白质和小分子的特征信息。但是表现性能逊于KDEEP、RF-Score的方法,这可能是由于我们的方法在构建矩阵时,为了防止矩阵的坐标越界,初始化矩阵的每个维度的边长都和数据集中所有原子矩阵所对应维度的边长的最大值有关,这使得大部分矩阵的非零数值都集中在三维矩阵的坐标的原点附近,其余部分有大量零值,虽然我们通过缩小矩阵来减小上述情况带来的负面效果,但是也会伴随着矩阵像素精度的较少。总之,我们提出的方法在所有方法中属于中等表现水平,有很大的优化空间。
5" 结" 论
预测药物靶点亲和力是新药研发的关键步骤,本文中我们提出了一种基于结构特征的深度学习方法,通过蛋白质和小分子的原子坐标信息分别网格化成两个特征矩阵,并根据原子类型的信息赋予矩阵数值。本文在PDBbind数据集上进行了实验,实验结果表明我们提出的方法在所有性能上都优于X-Score和cyScore这两种传统的函数评分方法,是一项有前景的深度学习方法。
参考文献:
[1] MULLARD A. New drugs cost US$2.6 billion to develop [J].Nature Reviews Drug Discovery,2014,13(12):877.
[2] ASHBURN T T,THOR K B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs [J].Nature Reviews Drug Discovery,2004:673-683.
[3] ROSES A D. Pharmacogenetics in Drug Discovery and Development: A Translational Perspective [J].Nature Reviews Drug Discovery,2008(10):807-817.
[4] BALLESTER P J,MITCHELL J B O. A Machine Learning Approach to Predicting Protein-Ligand Binding Affinity with Applications to Molecular Docking [J].Bioinformatics,2010,26(9):1169-1175.
[5] ÖZTÜRK H,ÖZGÜR A,OZKIRIMLI E. DeepDTA: Deep Drug-Target Binding Affinity Prediction [J].Bioinformatics,2018,34(17):i821-i829.
[6] NGUYEN T,LE H,QUINN T P,et al. GraphDTA: Predicting Drug-Target Binding Affinity with Graph Neural Networks [J].Bioinformatics,2021,37(8):1140-1147.
[7] JIMÉNEZ J,DOERR S,MARTÍNEZ-ROSELL G,et al. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks [J].Bioinformatics,2017,33(19):3036-3042.
[8] JIMÉNEZ J,SKALIC M,MARTINEZ-ROSELL G,et al. KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks [J].Journal of chemical information and modeling,2018,58(2):287-296.
[9] NAIR V,HINTON G E. Rectified Linear Units Improve Restricted Boltzmann Machines [C]//Proceedings of the 27th Interational Conferenice on Machinse Learring,Haifa:[s.n.],2010.
[10] WANG R X,FANG X L,LU Y P,et al. The PDBbind Database: Methodologies and Updates [J].Journal of Medicinal Chemistry,2005,48(12):4111-4119.
[11] WANG R X,LAI L H,WANG S M. Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction [J].Journal of Computer-Aided Molecular Design,2002,16(1):11-26.
[12] CAO Y,LI L. Improved Protein-Ligand Binding Affinity Prediction by Using a Curvature-Dependent Surface-Area Model [J].Bioinformatics,2014,30(12):1674-1680.
作者简介:邵允昶(1999—),男,汉族,山东青岛人,硕士研究生在读,研究方向:基于深度学习的药物靶点亲和力研究;张媛媛(1986—),女,汉族,山东德州人,副教授,博士研究生,研究方向:人工智能在药物发现中的应用;江明建(1991—),男,汉族,山东青岛人,讲师,博士研究生,研究方向:基于深度学习的蛋白质组学研究。