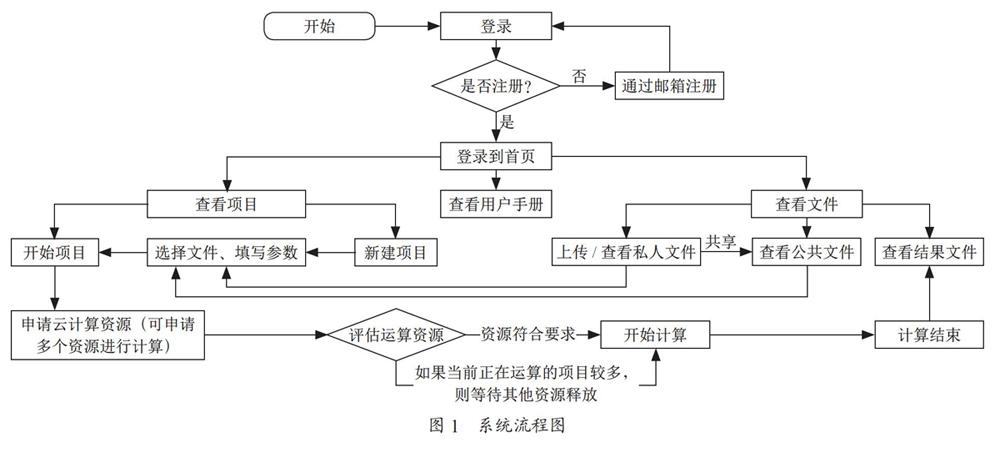
中图分类号:S664.2 文献标志码:A 文章编号:1002-2910(2025)02-0011-08
Analysis of the genetic characteristics of Castanea germplasms based on genome resequencing
WANG Jinping1, TIAN Shoule1, CAI Gongzhan2, SUN Xiaoli1, TAO Diyang³, SHEN Guangning1* (1.Shandong Institute of Pomology,Taian,Shandong 2710oo, China; 2.Shandong Shangnong Agricultural Technology Co.,Ltd.,Heze,Shandong,China;3.Culaishan Forest Farm ofTaian City,Taian, Shandong , China)
Abstract: In this study, genome resequencing technology was employed to conduct an analysis of the genetic characteristics of a total of 180 Castanea germplasms,which included Chinese chestnut (Castanea molissima) cultivars from 16 provinces in China, as wellas European chestnut (Castanea sativa) and Japanese chestnut (Castanea crenata). As a result, 48 374 108 genetic variation sites were successfully identified. According to the analysis of these variation sites,the l80 cultivars could be classified into eight major groups. There was a phenomenon of gene introgresson among different groups. The population dynamic changes of Chinese chestnut, European chestnut, and Japanese chestnut during the evolutionary process were significantly different. The study also discovered the gene flow of the Chinese chestnut populations from Beijing to Shandong. However, the study has limitations such as uneven distribution of the sample size and limited analysis dimensions. In the future, it is necessary to expand the sample scope and conduct in-depth research by integrating multi-omics approaches.
Key words: chestnut; germplasms; genome re-sequencing; genetic characteristics; population structure; gene flow
板栗(CastaneamollissimaBlume)在中国农林产业与生态建设中占据重要地位[1]。深入剖析其种质资源的遗传特性,是推动品种优化、培育高产优质且适应性强新品种的关键。以往关于板栗的遗传分析多采用形态学标记,通过观察植株的形态特征,如树形、叶形、果实形状等,以及基于简单序列重复(SSR)等分子标记技术。这些方法虽能初步揭示板栗种群间的遗传差异,但由于易受环境因素干扰,且所能检测到的遗传变异位点有限,无法全面、精确地展现板栗复杂的遗传全貌,满足现代遗传育种研究的需求[2]。
基因组重测序能够对已有参考基因组的物种进行深度测序,通过与参考基因组细致比对,能够高效、精准地识别出单核昔酸多态性(SNP)、短片段插入缺失(InDeI)以及大片段结构变异(SV)等丰富的遗传变异信息[3],助力解析群体遗传结构、挖掘关键基因,极大地推动了植物遗传育种进程[4]。
尽管基因组重测序技术在植物研究中成果丰硕,但栗属种质资源遗传特性分析方面的应用研究报道较少。本研究运用基因组重测序技术,对广泛收集的栗属种质资源展开全面深入的遗传分析,旨在明晰栗属种质资源的遗传多样性、明确群体结构划分以及梳理亲缘关系脉络,为栗属种质资源的保护利用和遗传改良提供理论与数据支撑。
1材料与方法
1.1 试验材料
本研究收集了180份来自不同地区的栗属种质资源(表1)。这些种质资源在形态特征、生态适应性等方面具有一定的差异,能够较好地代表栗属种质资源的多样性。
1.2 试验方法
1.2.1DNA提取与质量检测采用改良的CTAB法提取板栗叶片的基因组DNA,通过 0 . 7 5 % 琼脂糖凝胶电泳检测DNA片段大小和DNA降解程度;
NanoDropOne分光光度计检测DNA纯度,检测OD260/280比值在 1 . 8 ~ 2 . 2 ,无蛋白质和肉眼可见杂质污染;Qubit3.0荧光仪(LifeTechnologies,Carlsbad,CA,USA)检测DNA浓度,检测浓度大于 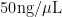 ,总量大于 2 μ g 。
,总量大于 2 μ g 。
1.2.2基因组重测序文库构建及库检DNA样品检测合格后,使用Covaris超声波破碎仪将DNA随机打断成 3 0 0 ~ 5 0 0 b p 的片段,打断后的样品磁珠进行片段选择,使得样品条带集中在 2 0 0 ~ 4 0 0 b p 左右。再经末端修复、加A尾、加测序接头、纯化、PCR扩增、PCR产物环化等步骤完成整个文库制备工作。
文库构建完成后,先使用Qubit2.0荧光定量仪进行初步定量,随后使用Agilent2100生物分析仪对文库的插入片段进行检测,插入片段大小符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量,以保证文库质量。测序时,采用双端150bp的测序策略,测序深度达到10X以上,以保证数据的准确性和可靠性。
DNBSEQ上机测序。检测合格文库安排上机测序(DNBSEQ):单链环状DNA分子通过滚环复制,形成一个包含300多个拷贝的DNA纳米球(DNB)。将得到的DNBs采用高密度DNA纳米芯片技术,加到芯片上的网状小孔内,通过联合探针锚定聚合技术(cPAS)进行测序。
1.2.3测序数据筛选测序平台得到的原始图像数据文件,经过碱基识别分析即可转化为原始测序序列,称为RawData或RawReads,对Rawreads过滤,得到Cleanreads。使用fastp[5]对原始数据进行质控与过滤,具体过滤标准如下:剔除序列中的接头序列,即adapter;去除reads尾部的polyG和polyX;以滑窗的方式统计窗口内的碱基计算平均质量值,将低质量的滑窗剪裁掉;剔除N个数大于5的reads;剔除质量低于15的碱基占比高于 40 % 的reads;剔除过滤后长度低于 1 5 b p 的reads。
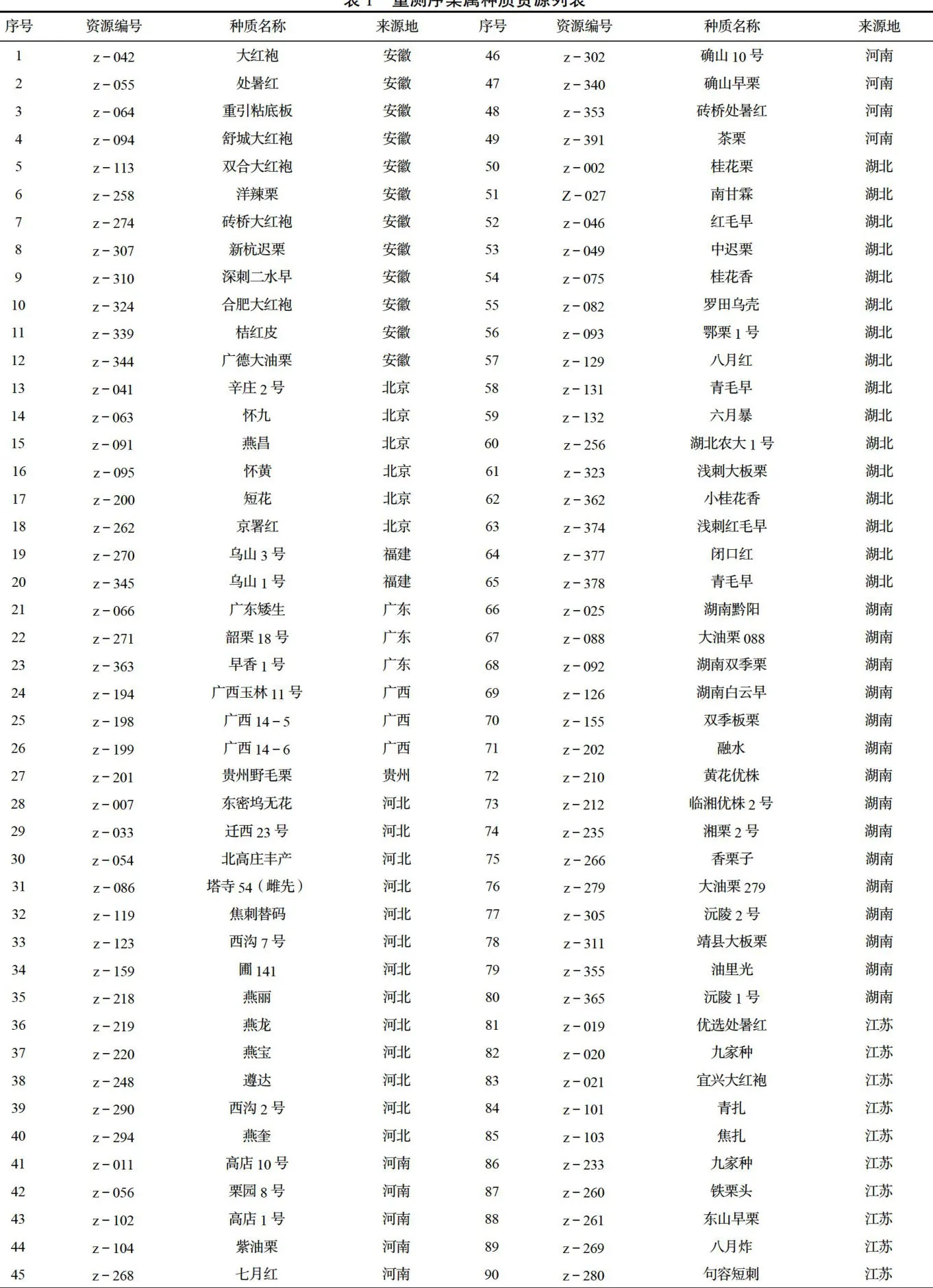 表1重测序栗属种质资源列表
表1重测序栗属种质资源列表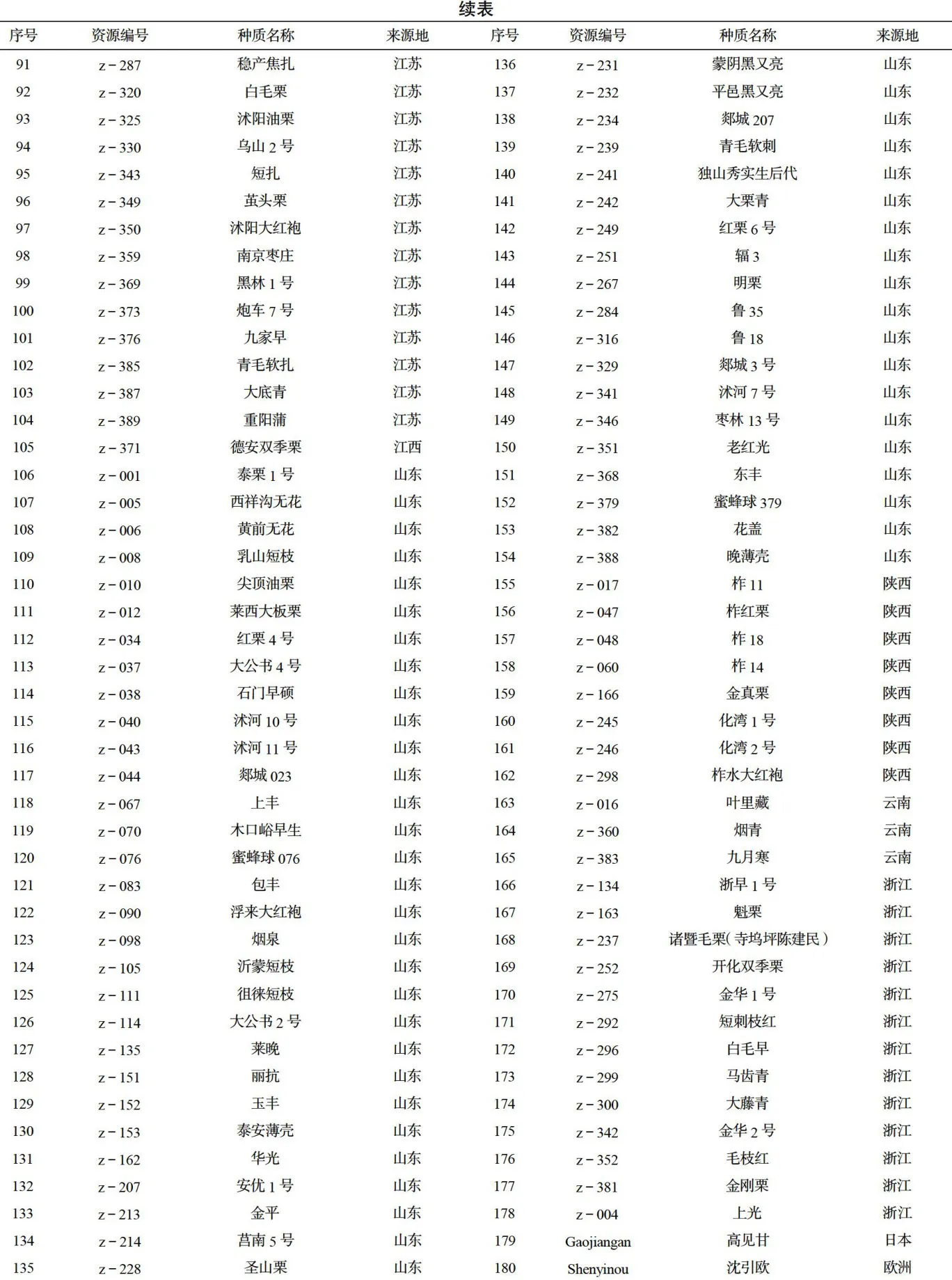 注:列表中有同一名称的资源,考虑到可能为同名异物,均作为研究对象。
注:列表中有同一名称的资源,考虑到可能为同名异物,均作为研究对象。
1.2.4变异鉴定利用生物信息学软件将测序数据与板栗参考基因组[(表2)进行比对。首先,使用BWA[7软件将测序reads比对到参考基因组上(参数:mem-R,其余参数采用软件默认参数),然后利用SAMtools[8软件对比对结果进行排序(参数:sort)、去重(参数:markdup-r)等处理,将比对结果从sam(SequenceAlignment/MAP)文件转为排序后的bam文件(binaryAlignment/Map),统计比对情况。为了更具体更细致的评估各样本比对参考基因组上的情况,采用滑窗的方式将基因组划分成大小相等的子区间,统计各个子区间内的平均测序深度(比对上该区间总的碱基数除以区间大小)以及覆盖度(区间内有reads覆盖的区域占区间大小的比例),从而更全面的了解各个样本的比对情况。
由于二代测序数据reads长度较短且基因组上存在着重复序列从而导致reads的错误比对,同时测序数据的分布不均匀。这些原因可能会导致变异鉴定结果的假阳性存在。因此为了降低这一比例,获得高质量的SNP和INDEL,使用GATK[9](版本:4.2.5.0;参数:VariantFiltration),根据GATK官方推荐的hard-filtering标准对鉴定到的SNP和Indel分别进行过滤,具体过滤标准如下:
基于GATKhard-filtering过滤后的剩下的变异结果文件,使用vcftools(版本:0.1.16;参数:-maf,-max-missing),剔除掉maf(次要等位基因频率)小于0.03,基因组型缺失比例大于 20 % 的位点。最后使用剩余的变异位点进行群体结构分析。
1.2.5群体结构分析使用PHYLIP[10](版本:3.696;参数:neighbor)中的邻接法(neighbor-joiningmethods,简称NJ),构建进化树。后续基于树文件(newick格式)使用ggtree进行可视化。
使用fastStructure[11,2](版本:1.0;参数:-K- seed - c v= 1 0 )进行群体遗传结构分析,K值取2~10,基于不同的seed值针对每个K值独立重复10次(最佳K值根据 fastStructure 提供的chooseK.py程序以及具体的群体背景进行确定)。
使用PSMC[13] (pairwise sequentially Markoviancoalescent)(版本:latest;参数: - N2 5 - t1 5 - r5 -b-p 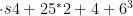 ,碱基突变速率:
,碱基突变速率: 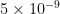 每代年数:15),利用个体重测序数据推测该个体所属的种群在历史上各个时期的有效群体大小。
每代年数:15),利用个体重测序数据推测该个体所属的种群在历史上各个时期的有效群体大小。
利用TreeMix[14]软件通过从多个种群中获得等位基因频率,返回该种群的最大似然(ML)树,并推断基因流。
2 结果与分析
2.1栗属种质资源统计
根据来源对重测序栗属资源进行了分析,180份栗属种质资源分别来源于中国16个省(市)以及欧洲和日本国家,其中山东49份、河北13份、安徽11份、湖南15份、湖北16份、江苏24份、浙江14份、河南9份、陕西8份、北京6份、云南3份、广东3份、广西3份、福建2份、江西1份、贵州1份、欧洲栗1份、日本栗1份(表1)。
2.2 测序数据质控
通过对180份栗属资源的测序,每个样本得到的Rawreads数量 6 3 0 1 0 8 6 6 ~ 1 8 8 5 7 2 2 0 4 ,通过进行质控与过滤获得Cleanreads的数量范围为: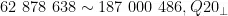 ratio在 9 7 . 2 3 % 以上,Q30ratio在 9 0 . 2 0 % 以上,GC含量 34 . 3 9 %~3 6 . 8 0 % 。以2020年发表的板栗全基因组组装结果为参考基因组,基因组大小为6 8 8 . 9 9 M b 。
ratio在 9 7 . 2 3 % 以上,Q30ratio在 9 0 . 2 0 % 以上,GC含量 34 . 3 9 %~3 6 . 8 0 % 。以2020年发表的板栗全基因组组装结果为参考基因组,基因组大小为6 8 8 . 9 9 M b 。
 表2参考基因组信息注:ContigNumber:1Kb以上scafold中间的contig数目;Contiglength(bp):为1Kb以上scafold中间的contig的长度;ContigN50(bp):contigN50的长度;Contig N90(bp):contig N90的长度;Contig max(bp):最长的contig的长度;GCcontent ( % ) ):GC含量。
表2参考基因组信息注:ContigNumber:1Kb以上scafold中间的contig数目;Contiglength(bp):为1Kb以上scafold中间的contig的长度;ContigN50(bp):contigN50的长度;Contig N90(bp):contig N90的长度;Contig max(bp):最长的contig的长度;GCcontent ( % ) ):GC含量。
通过对比对结果分析,180个样本的比对率在9 7 . 6 2 % ~ 9 9 . 4 6 % ,对参考基因组的覆盖度在
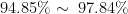 ,平均测序深度在13.11X~3 2 . 4 3 X 。
,平均测序深度在13.11X~3 2 . 4 3 X 。
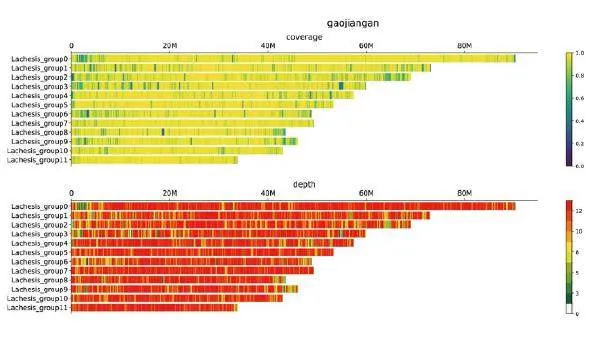 图1品种高见甘基因组内各窗口平均测序深度以及覆盖度注:Chrom:染色体编号;Start:窗口起始位置;End:窗口终止位置;Mean_depth:窗口内平均测序深度;Count:窗口mapping的reads数量;Coverage窗口覆盖的碱基数;Length:窗口长度;coverage_rate:窗口覆盖度。
图1品种高见甘基因组内各窗口平均测序深度以及覆盖度注:Chrom:染色体编号;Start:窗口起始位置;End:窗口终止位置;Mean_depth:窗口内平均测序深度;Count:窗口mapping的reads数量;Coverage窗口覆盖的碱基数;Length:窗口长度;coverage_rate:窗口覆盖度。
2.3等位基因类型及统计
基于碱基类型信息,对48374108个变异位点进行了分类,其中二等位变异、三等位变异、四等位变异、五等位变异、六等位变异、七等位变异数量分别为43564584、4015291、405120、130080、91839、167194。对每个品种具体的变异情况(INDEL变异、碱基转换、颠换、颠换转换比、纯合基因型、杂合基因型以及杂合比等)进行了统计。整体变异中各类型颠换以及转换变异的比例进行了可视化(图2),针对变异在基因组上的分布进行可视化(图3)。
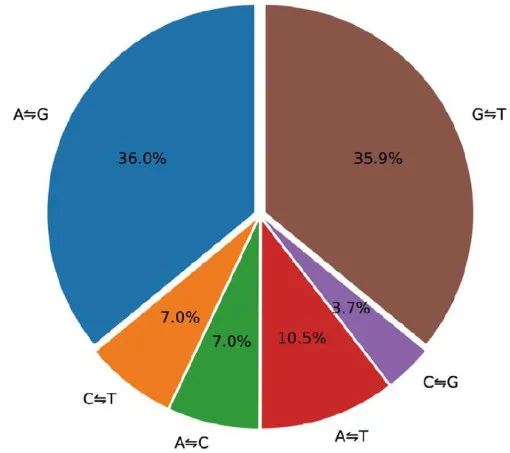 图2转换颠换比例图
图2转换颠换比例图
2.4群体结构分析
群体进化树构建。根据所测180份资源的重测序结果,基于邻接法构建了系统进化树,180份资源可以分成八大类群,大的聚类结果与品种地理来源没有明显相关。
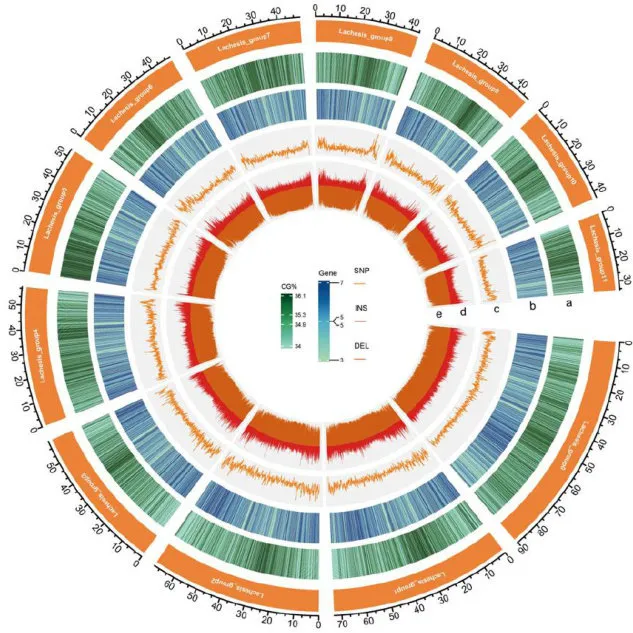 图1品种高见甘基因组内各窗口平均测序深度以及覆盖度图3全基因组变异分布注:a:CG含量;b:基因数量;c:SNP数量;d:Insertion数量;e:Deletion数量。
图1品种高见甘基因组内各窗口平均测序深度以及覆盖度图3全基因组变异分布注:a:CG含量;b:基因数量;c:SNP数量;d:Insertion数量;e:Deletion数量。
如来自于湖南的z-305(沅陵2号)、z-365(沅陵1号),江苏的z-280(句容短刺)、z-385(青毛软扎)、z-373(炮车7号)、Z-101(青扎)、z-103(焦扎),湖北的 z - 0 9 3 (鄂栗1号),山东的z-251(辐3)、z-239(青毛软刺)、z-284(鲁35)、z-341(沭河7号),河南的z-353(砖桥处暑红),安徽的z-042(大红袍)、z-094(舒城大红袍),河北的 z - 2 9 0 (西沟2号)等16个品种聚为一类。有部分小规模聚类值得关注,如来自广东的z-271(韶栗18号)、Z-363(早香1号)聚为一类;来自江苏的z-019(优选处暑红)与来自安徽的z-055(处暑红)聚到一起,来自湖北的z-046(红毛早)和z-374(浅刺红毛早)聚到一起;河北的 z - 0 0 7 (东密坞无花)与z-201(贵州野毛栗)聚到一起;z-198(广西14-5)、z-011(高店10号)、z-324(合肥大红袍)z-320(白毛栗)与Shenyinou(沈引欧)聚为一类。
群体Structure分析。基于检测过滤后的品种SNP数据,使用fastStructure进行群体遗传结构分析,当 K=8 时Cv-Error较小,与系统发育树分群结果相一致,各来源地表现出基因结构相互渗透的现象(图4)。因此认为收集的180份样本分为8个类群比较合适。
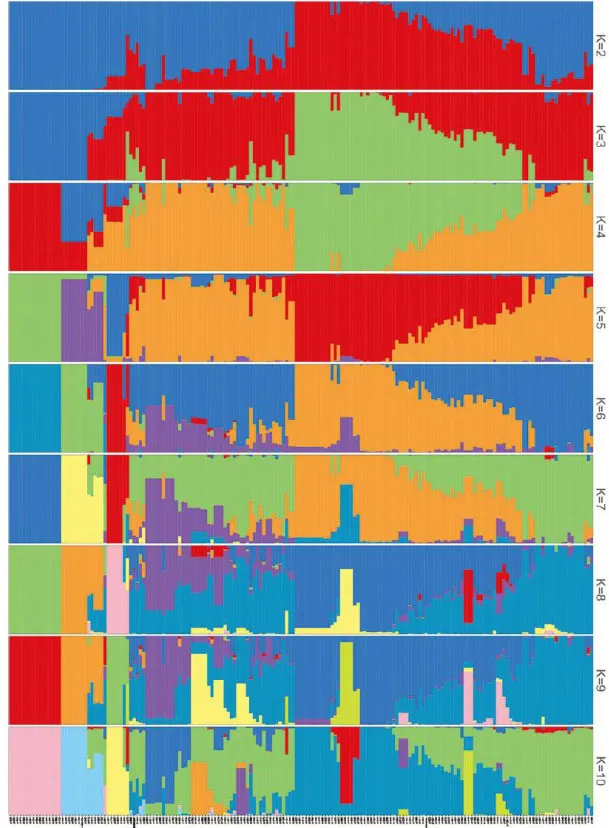 图4不同K值的群体结构图
图4不同K值的群体结构图
2.5 有效群体大小分析
本研究从山东、河北、河南、江苏、安徽、日本栗、欧洲栗等群体中分别选择1个样本的重测序数据推测个体所属的种群在历史上各个时期的有效群体大小。如图5所示,除广东外,中国各个产区的有效群体变化趋势相近,而日本栗有效群体变化趋势则与中国栗差异较大。由图可知,大约在1 Ma.B.P.(Million-anniversary Before Present)中国栗、日本栗、欧洲栗三个群体大小大致重合在一起。中国栗在 0 . 4 ~ 0 . 5 M a . B . P 有效群体达到最大值,日本栗在0.2~0.3Ma.B.P.有效群体达到最大值,且有效群体数量明显高于中国栗。此后经历了长期的衰退。大约在 2 0 ~ 2 5 K a .B.P.(kilion -anniversaryBeforePresent)又开始逐渐扩张。
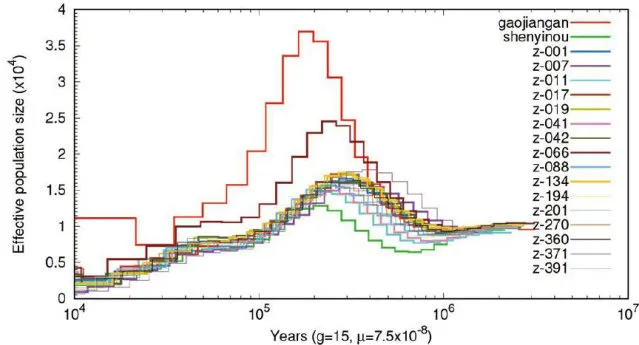 图5有效群体分析图
图5有效群体分析图
2.6 基因流分析
根据各群体中每个SNP类型变异位点等位基因数量统计,以每省(市)的品种为一个种群,使用Treemix进行基因流预测。由于广西、福建、广东等省份的品种数量较少,不进行分析。对湖北、湖南、山东、陕西、河北、北京、河南、安徽、浙江、江苏等10个省(市)的品种进行基因流分析。如图6所示,河南、安徽、浙江、江苏、湖北、湖南漂移参数较低,群体间遗传差异小;检测出从北京板栗种群向山东板栗种群的基因流,反映出山东早期从北京引种交流活动比较频繁。
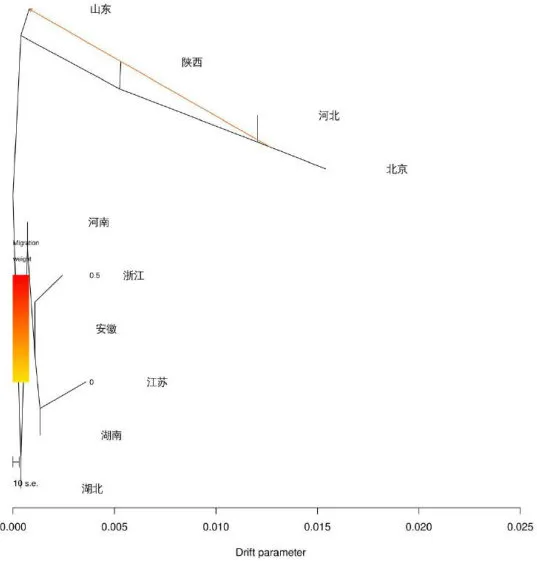 图6基因流分析图
图6基因流分析图
3小结与讨论
3.1群体结构分析在育种中的利用
本研究首次运用基因组重测序技术对来自中国、欧洲和日本共180份栗属种质资源展开了全面的遗传特性分析,成功获取了高质量的测序数据,并精准鉴定出丰富的遗传变异位点。
通过系统进化树和Structure等群体结构分析表明,180份资源可划分为八大类群,且各地区品种间存在明显的基因渗透现象,这充分证实了中国板栗资源遗传背景的多样性以及不同地区品种间频繁的基因交流。有效群体大小分析揭示了中国栗、日本栗和欧洲栗在进化历程中的群体动态变化,为深入理解板栗的进化历史提供了关键线索。基因流分析进一步解释了不同省份板栗群体间的遗传关系,为板栗品种的遗传改良提供了信息。
研究鉴定出的大量遗传变异位点,为后续开展板栗功能基因挖掘、分子标记开发、进化生物学研究以及深入解析板栗重要农艺性状的遗传机制提供了丰富的素材。
板栗在相互引种和选育过程中出现了同名异物或者同物异名的现象,如本研究通过分析发现优选处暑红与处暑红,红毛早与浅刺红毛早等很可能为同物异名。明确的群体结构和遗传关系,能够指导种质资源的合理收集、保存和管理,避免遗传资源的重复收集和流失。育种工作者可依据遗传距离和基因流信息,精准选择具有较大遗传差异的亲本进行杂交,从而提高育种效率,培育出更具优良性状的新品种,有力推动板栗产业的高质量发展。
3.2 研究展望
尽管本研究取得了一系列重要成果,但仍存在一定的局限性。在试验材料方面,虽然收集了来自多个地区的栗属种质资源,但欧洲栗、日本栗以及中国的广西、云南等部分地区的样本数量相对较少,可能对群体遗传结构分析的准确性产生一定影响。未来研究可进一步扩大样本收集范围,增加样本数量,以更全面地揭示栗属种质资源的遗传多样性。
在数据分析方面,本研究主要聚焦于群体结构、有效群体大小和基因流等常规分析,后续研究将结合表型数据,运用全基因组关联分析(GWAS)等方法,深入挖掘与板栗产量、品质、抗病性等重要性状紧密相关的遗传标记和功能基因,为分子标记辅助育种提供更精准的靶点。
此外,本研究仅涉及基因组层面的分析,而植物的生长发育和性状表现是一个复杂的调控网络,受到转录组、蛋白质组和代谢组等多个层面的协同调控。未来将结合开展多组学联合分析,多个维度解析板栗的遗传调控机制,为栗属种质资源的创新利用和遗传改良拓宽路径。
参考文献:
[1]阚黎娜,李倩,谢爽爽,等.我国板栗种质资源分布及营养成分比较[J].食品工业科技,2016,37(20):396-400.
[2]李沛,何治霖,谈月霞,等.基于重测序数据与表型性状的宽皮柑橘遗传多样性分析与优异种质筛选[J].中国农业科学,2024,57(23):4761-4795.
[3]姚远,邓利君,胡娟,等.脆红李及其早熟芽变全基因组重测序分析[J].园艺学报,2024,51(10):2255-2266.
[4]VARSHNEYRK,GRANERA,SORRELLSME.Genomicsequencing and discovery of markers for crop improvement[J].Trends in biotechnology,2009,27(7):442-450.
[5]CHEN S, ZHOU Y,CHEN Y,et al. Fastp: an ultra- fast all- in-oneFASTQ preprocessor[J].Bioinformatics,2018,34(17):1884 -1890.
[6]WANG J, TIAN S,SUN X,et al. Construction of Pseudomoleculesfor the Chinese Chestnut (Castanea mollissima) Genome[J].G3-Genes GenomesGenetics,2020,10(10):3565-3574.
[7]HENG L, RICHARD D. Fast and accurate short read alignment withBurrows-Wheeler transform[J].Bioinformatics,2010,14(25):1754-1760.
[8]LI H,HANDSAKER B,WYSOKER A,et al.The SequenceAlignment/Map format and SAMtools[J].Bioinformatics,2009,25(16):2078-2079.
[9]DANECEK P,AUTONA,ABECASIS G,et al.The variant allformatandVCFtools[J].Bioinformatics,201l,27(15):2156-2158.
[10]RAJA,STEPHENS M,PRITCHARD JK. Fast STRUCTURE:Variational Inference of Population Structurein Large SNPDataSets[J].Genetics,2014,197(2):573-589.
[11]LAM HM, XU X, LIU X,et al. Addendum: Resequencing of 31wild and cultivated soybean genomes identifies paterns of geneticdiversity and selection[J].Nature Genetics,2011,43(4):387.
[12]LAYER R M,CHIANGC,QUINLAN AR,et al. LUMPY:aprobabilisticframeworkforstructuralvariantdiscovery[J].GENOMEBIOLOGY,2014,15(6):1-19.
[13]SHENGLIN,LIU,MICHAEL,et al. PSMC (pairwise sequentiallyMarkovian coalescent) analysis of RAD (restriction site associatedDNA) sequencing data[J]. Molecular Ecology Resources,2017,(17):631-641.
[14]FITAK R R. Opt M: estimating the optimal number of migrationedges on population trees using Treemix[J].Biology Methods andProtocols,2021,6(1):1-10.